The representation on a phylogenetic tree of the divergence of two or more taxa from a common ancestor. A branch point is usually shown as a dichotomy in which a branch representing the ancestral lineage splits (at the branch point) into two branches, one for each of the two descendant lineages.
What is a phylogenetic tree and how to construct it?
What is the Phylogenetic Tree?
- Construction of the Phylogenetic tree. There are two different methods based on which the phylogenetic tree is constructed. ...
- Steps for preparing the Phylogenetic Tree
- Types of Phylogenetic Trees. Make the inference about the most common ancestor of the leaves or branches of the tree. ...
- Importance of Phylogenetic Tree. ...
How to construct a phylogenetic tree?
Protocol
- Acquiring the Sequences. Ironically, the first step is the most intellectually demanding, but it often receives the least attention.
- Aligning the Sequences. If the Alignment Explorer window is not already open, in MEGA5's main window choose Open a File/Session from the File menu.
- Estimate the Tree. ...
- Present the Tree. ...
How to analyze phylogenetic tree?
How to construct a Phylogenetic tree ?
- Phylogenetic Analysis and the Role of Bioinformatics. Molecular data that are in the form of DNA or protein sequences can also provide very useful evolutionary perspectives of existing organisms because, ...
- Steps in Phylogenetic Analysis. ...
- Bioinformatics Tools for Phylogenetic Analysis. ...
- References. ...
How to read a phylogram tree?
• The tips of the phylogenetic tree represent groups of descendent taxa (often species) • The internal nodes of the tree represent the common ancestors of those descendents. • The tips are the present and the internal nodes are the past. • The edge lengths in some trees correspond to time estimates – evolutionary time.
What does a branch point in a phylogenetic tree represent quizlet?
What does a branch point in a phylogenetic tree represent? A branch point represents a point at which two evolutionary lineages split from a common ancestor.
What best describes a branch point in a phylogenetic tree?
The point where a split occurs in a tree, called a branch point, represents where a single lineage evolved into distinct new ones. Many phylogenetic trees have a single branch point at the base representing a common ancestor of all the branches in the tree.
What does a branching point indicate?
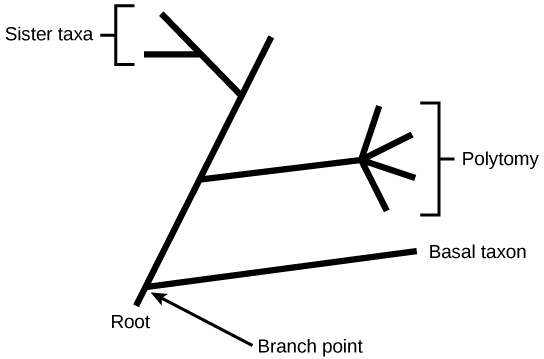
A branch point indicates where two lineages diverged. A lineage that evolved early and remains unbranched is a basal taxon. When two lineages stem from the same branch point, they are sister taxa. A branch with more than two lineages is a polytomy.
What do branch points and lines represent in a phylogenetic tree?
Key Points Branch points in a phylogenetic tree represent a split where a single lineage evolved into a distinct new one, while basal taxon depict unbranched lineages that diverged early from the root. Unrooted trees portray relationships among species, but do not depict their common ancestor.
What is branch point in biology?
A single metabolite that is an intermediate in two or more biosynthetic pathways, e.g. pyruvate (a precursor of acetyl-CoA, alanine and oxaloacetate), chorismic acid (a precursor of phenylalanine, tryptophan and tyrosine). Tags: Molecular Biology.
What do the branches on the tree tell you about the relationships among different species?
Unrooted trees don't show a common ancestor but do show relationships among species. In a rooted tree, the branching indicates evolutionary relationships (Figure 3). The point where a split occurs, called a branch point, represents where a single lineage evolved into a distinct new one.
What is branch cut and branch point?
A branch cut is a curve in the complex plane such that it is possible to define a single analytic branch of a multi-valued function on the plane minus that curve. Branch cuts are usually, but not always, taken between pairs of branch points.
Which branch point represents the most recent common?
branch point 1This phylogenetic tree is rooted. The tree includes the most recent common ancestor of all living species of bears (branch point 1).
What is a node on a phylogenetic tree?
Nodes are the points at the ends of branches which represent sequences or hypothetical sequences at various points in evolutionary history.
What are the two types of phylogenetic trees?
Adopting the perspective of graph theory, Martin et al. (2010) described two kinds of phylogenetic trees, which they termed node-based and stem - or branch-based , that differ with respect to the biological interpretations of their component nodes and branches. After establishing equivalency between the two different kinds of trees in terms of encoded information regarding taxa and their phylogenetic relationships, Martin et al. (2010) argued that node-based names should be applied only in the context of node-based trees, and that branch-based names should be applied only in the context of branch-based trees, because node-based names cannot exist on branch-based trees and vice versa. They also suggested that the International Code of Phylogenetic Nomenclature or PhyloCode ( Cantino and de Queiroz 2010) confuses the two kinds of names and trees and should therefore be amended to adopt one or the other of the two kinds of trees and the corresponding kind of names.
What is the earliest ancestor of 7?
Here, the name is defined as designating the clade originating in the earliest ancestor of 7 that is not an ancestor of 10. The earliest ancestor of 7 that is not an ancestor of 10 is 1, so the designated clade contains 1 and all of its descendants (2–9).
What is graph theoretic perspective?
A graph theoretic perspective is useful for clarifying issues concerning both the representation of phylogeny using trees (minimal ly connected graphs) and methods for applying taxon names in the context of such trees ( de Queiroz 2007; Martin et al. 2010 ). Consideration of alternative significations of the nodes and branches of phylogenetic trees suggests that the terminology applied to different kinds of phylogenetic trees by Martin et al. (2010), as well as that applied to different kinds of phylogenetic definitions in the PhyloCode ( Cantino and de Queiroz 2010 ), are ambiguous or potentially confusing. Nevertheless, both minimum-clade (“node-based”) and maximum-clade (“branch-based”) definitions are not only applicable in the context of both relationship (“node-based”) and lineage (“branch-based”) trees, both kinds of phylogenetic definitions are also necessary components of a nomenclatural system that must necessarily function in the context of incomplete taxon sampling and changing hypotheses about phylogeny.
Is phylocode a single tree?
Martin et al. (2010, p. 10) proposed that the PhyloCode adopt only a single kind of tree and the corresponding kind of definition. I have demonstrated that their proposal was based on an incorrect proposition that “node-based” names are incompatible with “branch-based” trees and vice versa. Nonetheless, given that minimum-clade (“node-based”) and maximum-clade (“branch-based”) definitions are used to apply names to entities of the same fundamental kind (clades), and that both kinds of definitions can be applied in the context of both relationship (“node-based”) and lineage (“branch-based”) trees, one might still be tempted to conclude that a single kind of definition is sufficient. At least for people who equate the ancestors specified by phylogenetic definitions with entire species (rather than parts of species), it might seem that minimum- and maximum-clade definitions are simply different methods for specifying the same clades and ancestors ( Fig. 4) (see Frost and Kluge 1994; Sereno 1999 ). Such a conclusion, although correct in the context of complete and error-free knowledge of phylogeny, is over-simplified in the context of real (i.e., incomplete and at least potentially inaccurate) phylogenetic hypotheses, which is to say that there are pragmatic reasons for using both kinds of definitions even in the context of a single kind of tree. In the rest of this section, I will assume, for the sake of simplifying the discussion, that the ancestor in which a clade originates is an entire species.
Why are long branches attracted to the base of a phylogenetic tree?
Long branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched.
What is LBA in phylogenetics?
In phylogenetics, long branch attraction ( LBA) is a form of systematic error whereby distantly related lineages are incorrectly inferred to be closely related. LBA arises when the amount of molecular or morphological change accumulated within a lineage is sufficient to cause that lineage to appear similar (thus closely related) to another long-branched lineage, solely because they have both undergone a large amount of change, rather than because they are related by descent. Such bias is more common when the overall divergence of some taxa results in long branches within a phylogeny. Long branches are often attracted to the base of a phylogenetic tree, because the lineage included to represent an outgroup is often also long-branched. The frequency of true LBA is unclear and often debated, and some authors view it as untestable and therefore irrelevant to empirical phylogenetic inference. Although often viewed as a failing of parsimony-based methodology, LBA could in principle result from a variety of scenarios and be inferred under multiple analytical paradigms.
Why is LBA problematic?
LBA was first recognized as problematic when analyzing discrete morphological character sets under parsimony criteria, however Maximum Likelihood analyses of DNA or protein sequences are also susceptible. A simple hypothetical example can be found in Felsenstein 1978 where it is demonstrated that for certain unknown "true" trees, some methods can show bias for grouping long branches, ultimately resulting in the inference of a false sister relationship. Often this is because convergent evolution of one or more characters included in the analysis has occurred in multiple taxa. Although they were derived independently, these shared traits can be misinterpreted in the analysis as being shared due to common ancestry.
What is the result of LBA in evolutionary analysis?
The result of LBA in evolutionary analyses is that rapidly evolving lineages may be inferred to be sister taxa, regardless of their true relationships. For example, in DNA sequence-based analyses, the problem arises when sequences from two (or more) lineages evolve rapidly. There are only four possible nucleotides and when DNA substitution rates are high, the probability that two lineages will evolve the same nucleotide at the same site increases. When this happens, a phylogenetic analysis may erroneously interpret this homoplasy as a synapomorphy (i.e., evolving once in the common ancestor of the two lineages).
What is a hypothetical example of a false sister relationship?
A simple hypothetical example can be found in Felsenstein 1978 where it is demonstrated that for certain unknown "true" trees, some methods can show bias for grouping long branches, ultimately resulting in the inference of a false sister relationship.
What is Hennig's Auxiliary Principle?
Hennig's Auxiliary Principle suggests that synapomorphies should be viewed as de facto evidence of grouping unless there is specific contrary evidence (Hennig, 1966; Schuh and Brower, 2009). A simple and effective method for determining whether or not long branch attraction is affecting tree topology is the SAW method, named for Siddal and Whiting. ...
Why is convergent evolution misinterpreted?
Although they were derived independently, these shared traits can be misinterpreted in the analysis as being shared due to common ancestry.